Cannabinoid Ethers: Identification, Binding Interactions, and their ADME Properties
This review highlights an in-silico assessment of the binding interactions of pro-drug moiety analogs and an importance in identifying potential issues that would need further testing to ensure safety for consumers such as the hERG values which are pertinent in the pharmaceutical fields in whether or not drugs can be safe to use.
New cannabinoid analogs are constantly being synthesized and marketed to the public as ways to increase psychotropic activity or to remain competitive within the market. The issue with new analogs is the lack of proper testing and characterization—companies are misrepresenting what they sell, and customers are consuming unknown research compounds. Our group performed in-silico docking in CB1 and CB2 receptors, drug metabolism, and pharmacokinetics (DMPK) studies of methyl-, ethyl-, and pivaloy- ether cannabinoids analogs. A dataset of eight cannabinoid ethers was docked to the model to further analyze key interacting residues on the CB1 and CB2 receptors. In addition, the methyl ether derivatives of Delta 8-tetrahydrocannabinol (D8-THC) and hexahydrocannabinol (HHC) were synthesized and the full nuclear magnetic resonance (NMR) characterization of these compounds was achieved.



1. Introduction
Within the volatile cannabinoid market, the need to stay relevant and competitive grows: new cannabinoid derivatives are being introduced to circumvent regulations or to increase efficacy, with safety being called to attention. Pre-clinical testing of compounds can be expensive and without the correct staff to interpret and present results, can make the data null. Computer-aided in-silico techniques that can perform ADME (absorption, distribution, metabolism, and excretion) computation (1-4), can assist in cost-effective analysis to generalize indicative characteristics of the tested compounds to pose health hazards (5). Some popular cannabinoids such as Delta 8-THC, Delta 9-THC (D9-THC), and HHC are popping up with prodrug moieties, which include methyl, ethyl, and pivaloyl (PV) prodrug moieties being attached to the essential hydroxy group within the aromatic ring that contributes to the important cannabimimetic activity (6). The prodrug moieties are typically used to create a stronger “psychedelic high” specifically the -O acetate (7), which does not increase smokable highs but might delay metabolism and survive the first pass leading to increased “high” orally, though nothing substantial. The motif is followed through the changing of the groups, which can enact different effects, as well as delay metabolism increasing and modulating the “high” effects. Cannabinoid derivatives in which the hydroxyl group in the resorcinol core was removed or replaced by an alkyl chain to generate an ether group significantly decreased ligand binding to CB1 exhibiting better selectivity for the CB2 receptor. Phenol hydroxy (PH) is believed to participate in the hydrogen bonding interaction with the CB1 cannabinoid receptor (8). For example, tested dimethyl compounds such as 6,6,9-trimethyl-3-(2-methylhexan-2-yl)-6a,7,10,10a-tetrahydro-6H-benzo[c]chromen-1-ol (1-Figure 1) has significant affinities for both CB1 and CB2 receptor, showing 0.83 nM and 0.49 nM as affinity constant (Ki) of CB1 and CB2, respectively. However, its corresponding methyl ether analog, 1-methoxy-6,6,9-trimethyl-3-(2-methylhexan-2-yl)-6a,7,10,10a-tetrahydro-6H-benzo[c]chromene (2-Figure 1) presented high CB2 selectivity with Ki (CB1) >20,000 and Ki (CB2) =19 nM (8,9). This led us to investigate several other linkers on the market attached to HHC and THC scaffolds as the primary focus due to their upcoming popularity within the market and identify their binding relationships as well as their ADME characteristics to help shine a light on an ever-changing market and provide a semblance of information on potential toxicities if any present.
2. Materials and Methods
General Remarks: All commercial acids and solvents were American Chemical Society (ACS) and high-performance liquid chromatography (HPLC) grade, respectively. Acids and solvents were all purchased from Sigma Aldrich and were used without further purification. Cannabidiol (CBD) isolate was purchased in bulk from GVB Biopharma. Quantification was performed using high-performance liquid chromatography (HPLC) on an Agilent 1100 series equipped with Next Leaf CBX for Potency C18 RP column 2.7uM, 150x4.6mm. MeOH with CH2O2:H20 with CH2O2 is used for quantification of reactions. Reagents and solvents were ACS and HPLC grade, respectively. Reagents and solvents were all purchased from Sigma Aldrich and were used without further purification. CBD isolate was purchased in bulk from Columbia Basin Bioscience, OR. Quantification was performed using high-performance liquid chromatography (HPLC) on an Agilent 1100 series equipped with Next Leaf CBX for Potency C18 RP column 2.7uM, 150x4.6mm. MeOH with CH2O2:H20 with CH2O2 is used for quantification of reactions. Computation chemistry was performed according to methodology from Cruces et al. (10).
2.1 Proteins and Ligands Preparation
All molecular docking experiments were achieved on CybertronPC CLX 13th Gen Intel(R) Core(TM) i9-13900KF at 3.00 GHz comprising 24 computing cores. Schrödinger Release 2023-3: Glide software was used as the docking program (11,12). Crystal structures of CB1, CB2, were retrieved from the RCSB Protein Data Bank. CB1 [(PDB: 7V3Z]/, CB2 [(PDB: 5ZTY)]. The proteins were prepared using a protein preparation workflow tool on Schrödinger Protein Preparation Wizard (3,4). The external water molecules and ions were removed. Polar Hydrogens were added. Missing side chains were filled using Epic and PROPKA. Het states were generated at pH 7.4 (+/- 2.0). Heavy atoms converged to RMSD 0.30Å. 3D structures of cannabinoids and hydrogenated cannabinoids were established in 2D sketcher which was then exported as an SDF file and imported and prepared using LigPrep, to form 3D conformers, including the various 3D chiral conformations.
2.2 In Silico Molecular Docking
The grid parameter was generated covering the CB1 pockets for (PDB:7V3Z) [-42.91, -163.58, 306.7] for x,y,z coordinates. The ligand diameter midpoint box follows a 10Å x 10Å x 10Å x,y,z dimension. The grid parameter was generated covering the CB2 pockets for (PDB:5ZTY) [9.09, -0.17, -55.72] for x,y,z coordinates. The ligand diameter midpoint box follows a 10Å x 10Å x 10Å x,y,z dimension.
2.3. Prediction of ADMET Properties
The absorption, distribution, metabolism, excretion, and toxicity (ADMET) (13) properties of the cannabinoids were performed using QikProp version 4.4 integrated into Maestro (Schrödinger, LLC, New York, 2015) which predicts the widest variety of pharmaceutically relevant properties: QPlogS (predicted aqueous solubility), QPlogHERG (Predicted IC50 value for blockage of HERG K+ channels), QPPCaco (predicted apparent Caco-2 cell permeability. Caco-2 cells are a model for the gut-blood barrier), QPlogBB (predicted brain/blood partition coefficient), and % Human Oral Absorption (predicted human oral absorption in gastrointestinal tract on 0 to 100% scale). The calculated physicochemical descriptors are displayed in Table 4-SI. QikProp bases its predictions on the full 3D molecular structure and the global minimum energy conformer of each compound was used as input for ADMET properties.
2.4. Characterization Data
For the NMR analysis, the synthesized compounds were dissolved in CDCl3 (99.8 % atom D; Cambridge Isotope Laboratories, Andover, MA) and then transferred to a normal NMR tube. The proton and carbon nuclear magnetic resonance (1H-NMR and 13C-NMR, respectively) spectra were recorded using on a 500 MHz Bruker AVANCE II system at 25°C. 1H and 13C data sets were analyzed to yield complete 1H and 13C peak assignments. Coupling constants (J) are given in Hertz (Hz), and spin multiplicities are shown by the following symbols: s (singlet), d (doublet), dd (doublet of doublets), td (triplet of doublets), dt (doublet of triplets), dq (doublet of quartets), tt (triplet of triplets), and m (multiplet).
3. Experimental
General Procedure for the Methylation of Cannabinoids
To a solution of NaH 60% in mineral oil (152 g, 3.79 mol) in dry dimethylformamide (DMF)(2000 ml) cooled in an ice bath was added the corresponding cannabinoid derivative (3.1 mol) in DMF (2000 ml) dropwise as the reaction is very exothermic. The reaction was stirred in the ice bath until bubbles subsided, and then methyl iodide (236 ml, 3.79 mol) was added, stirring overnight at room temperature. HPLC after 18h showed no starting material. The reaction was quenched with water and extracted with dichloromethane (DCM). The organic layer was washed five times with water, dried over sodium sulfate, and concentrated in vacuo to give a dark red oil. The oil was purified by silica gel plug with hexanes: EtOAc (95:5) to afford the cannabinoid methyl ether.
1-methoxy-6,6,9-trimethyl-3-pentyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromene methyl ether (3)
Yellow oil. 938 g (92% of yield).1H NMR (400 MHz, CDCl3) δ (ppm): 6.31 (1H, d, J=1.7 Hz, H2); 6.25 (0.70H, d, J=1.7 Hz, H4, R-HHC-Me: 70%); 6.23 (0.30H, d, J=1.7 Hz, H4, S-HHC-Me: 30%); 3.80 (2.04H, s, OCH3_8’-R-HHC-Me: 70%); 3,78 (0.94H, s, OCH3_8’, S-HHC-Me: 30%); 3.02 (0.30H, dt, J=2.8 Hz, 11.0 Hz, H10a, R-HHC-Me: 70%); 2.86 (0.30H, dq, J=1.5 Hz, 2.8 Hz, 11.2 Hz, H10a, S-HHC-Me: 30%); 2.65 (0.30H, td, J=2.8 Hz, 11.2 Hz, H6a, S-HHC-Me: 30%); 2.52 (2H, td, J=2.8 Hz, 8.2 Hz, H1’); 2.43 (0.70H, td, J=2.8 Hz, 11.0 Hz, H6a, R-HHC-Me: 70%); 1.88-1.82 (1H, m, H10-1); 1.66-1.59 (3H, m, H9, H8-1, H7-1); 1.51-1.42 (1H, m, 10-2); 1.40-1.31 (8H, m, H7-2, H8-2, H2’, H3’, H4’); 1.15-1.07 (6H, m, CH3_6’, CH3_7’); 0.97-0.89 (6H, m, CH3_5’, CH3_9’). 13C NMR (100 MHz, CDCl3) δ (ppm): 159.1 (C1); 154.6 (C5); 142.5 (C3); 111.8 (C5a); 110.3 (C4); 103.2 (C2); 76.9 (C6); 55.8 (OCH3_8’); 49.4 (C6a); 39.3 (C10a); 36.1 (C10); 35.8 (C1’); 33.0 (C8); 31.8 (C2’); 30.9 (C3’); 29.7 (C9); 28.3 (C7); 27.9 (C4’); 22.8; 22.7 (CH3_6’, CH3_7’); 19.1 (CH3_9’); 14.2 CH3_5’).


1-methoxy-6,6,9-trimethyl-3-pentyl-6a,7,8,10a-tetrahydro-6H-benzo[c]chromene (5)
Yellow oil. 915.1 g (90 % yield). 1H NMR (400 MHz, CDCl3) (ppm): 6.32 (1H, d, J=1.7 Hz, H2); 6.26 (1H, d, J=1.7 Hz, H4); 5.43 (1H, dd, J=4.8 Hz, 9.0 Hz, H10); 3.81 (3H, s, OCH3_8’); 3.16 (1H, dt, J=4.1 Hz, 11.0 Hz, 9.0 Hz, H10a,); 2.67 (1H, td, J=4.8 Hz, 11.0 Hz, H6a); 2.51 (2H, td, J=2.7 Hz, 8.2 Hz, H1’); 2.18-2.10 (1H, m, H8-1); 1.90-1.81 (1H, m, H8-2); 1.80-1.77 (2H, m, H7-1, H7-2); 1.71 (3H, s, CH3_9’); 1.61 (2H, tt, J=2.2 Hz, 8.2 Hz, H2’); 1.37; 1.10 (3H, s, CH3_6’, CH3_7’); 1.35-1.33 (4H, m, H3’, H4’); 0.90 (3H, s, CH3_5’). 13C NMR (100 MHz, CDCl3) δ (ppm): 159.1 (C1); 154.5 (C5); 142.6 (C3); 135.1 (C9); 119.4 (C10); 112.0 (C5a); 110.4 (C4); 103.2 (C2); 76.6 (C6); 55.3 (OCH3_8’); 45.2 (C6a); 36.4 (C10a); 36.2 (C1’); 32.0 (C8); 31.8 (C2’); 30.9 (C3’); 28.1 (C7); 27.7 (C4’); 23.7; 22.7 (CH3_6’, CH3_7’); 18.6 (CH3_9’); 14.2 CH3_5’).
4. Results and Discussion
4.1. Molecular Docking of Cannabinoid Ethers
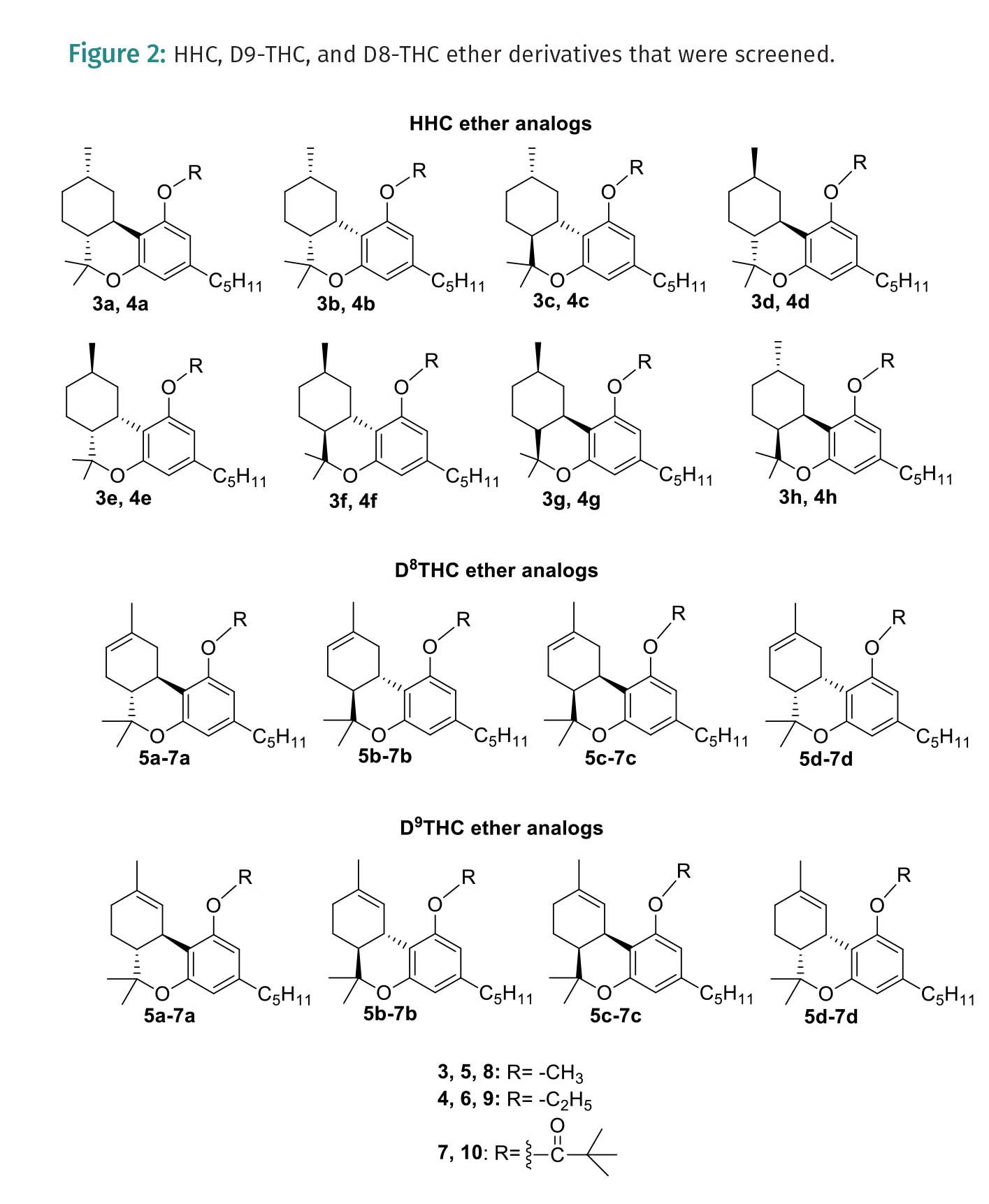
Our team used a virtual screen of eight cannabinoid ethers to explore the binding interaction and binding energies between cannabinoid ethers and CB1 (7V3Z) and CB2 (5ZTY) receptors. The compounds that were screened included HHC-OMe (3), HHC-ethyl ether (4), D8THC-OMe (5), D8THC-ethyl ether (6), D8THC-Pivaloyl-ether (7), D9THC- OMe (8), D9THC-ethyl ether (9), D9THC-Pivaloyl-ether (10), and their diastereomers (Figure 2). Using Jaguar to perform minimizations and calculate the density functional theory (DFT) for given scaffolds, the then minimized scaffolds were docked within 7V3Z and 5ZTY proteins that were prepared using the Schrödinger protein preparation workflow.


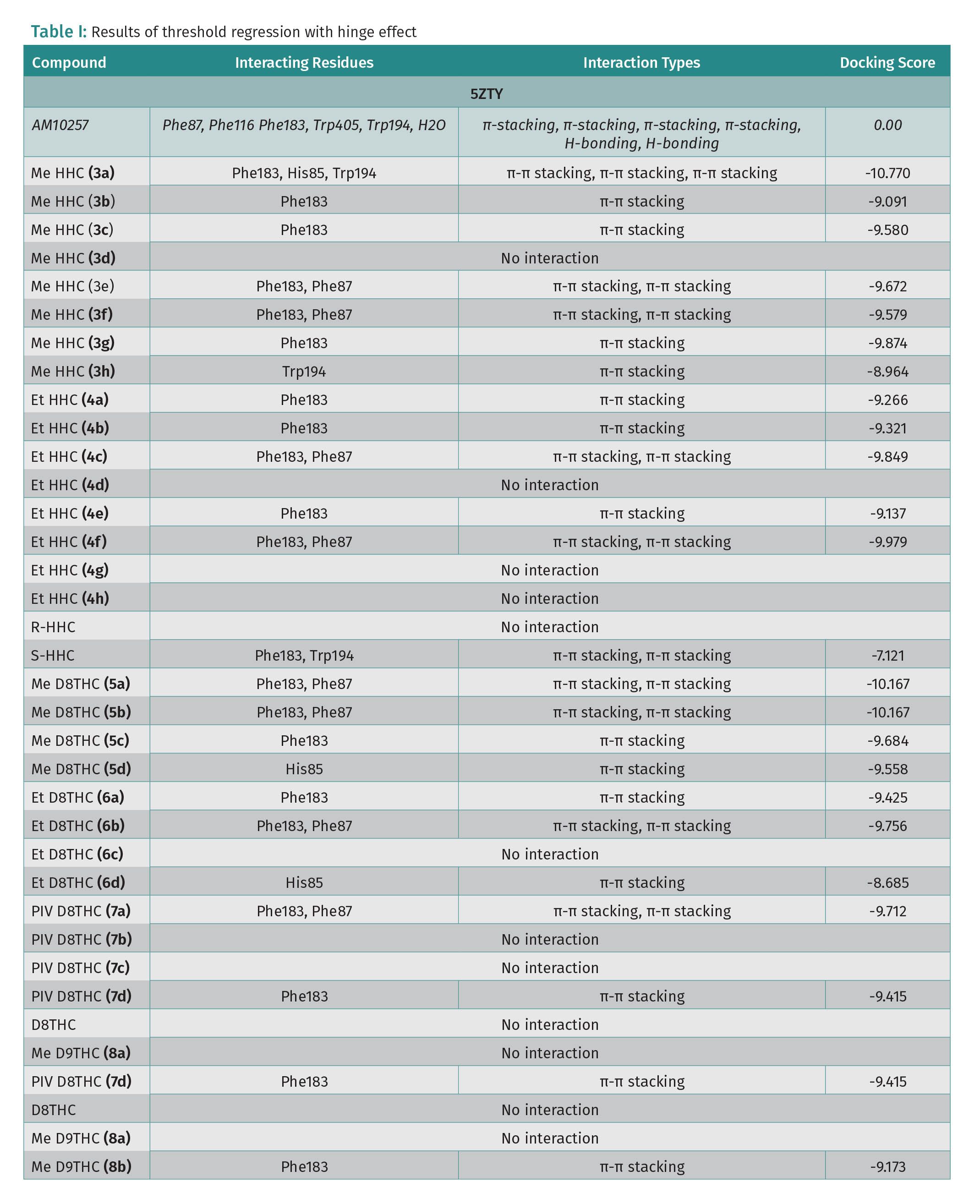


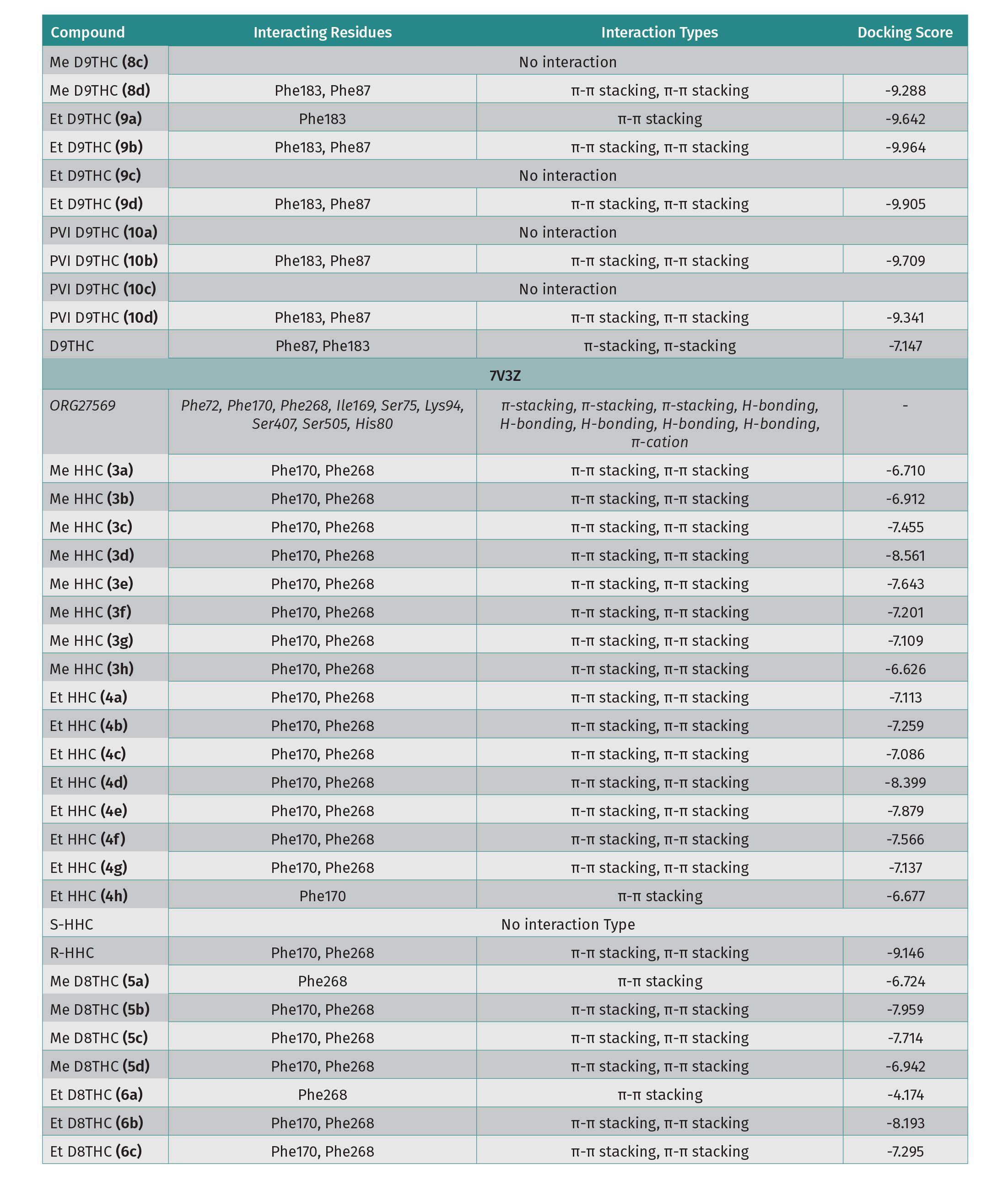


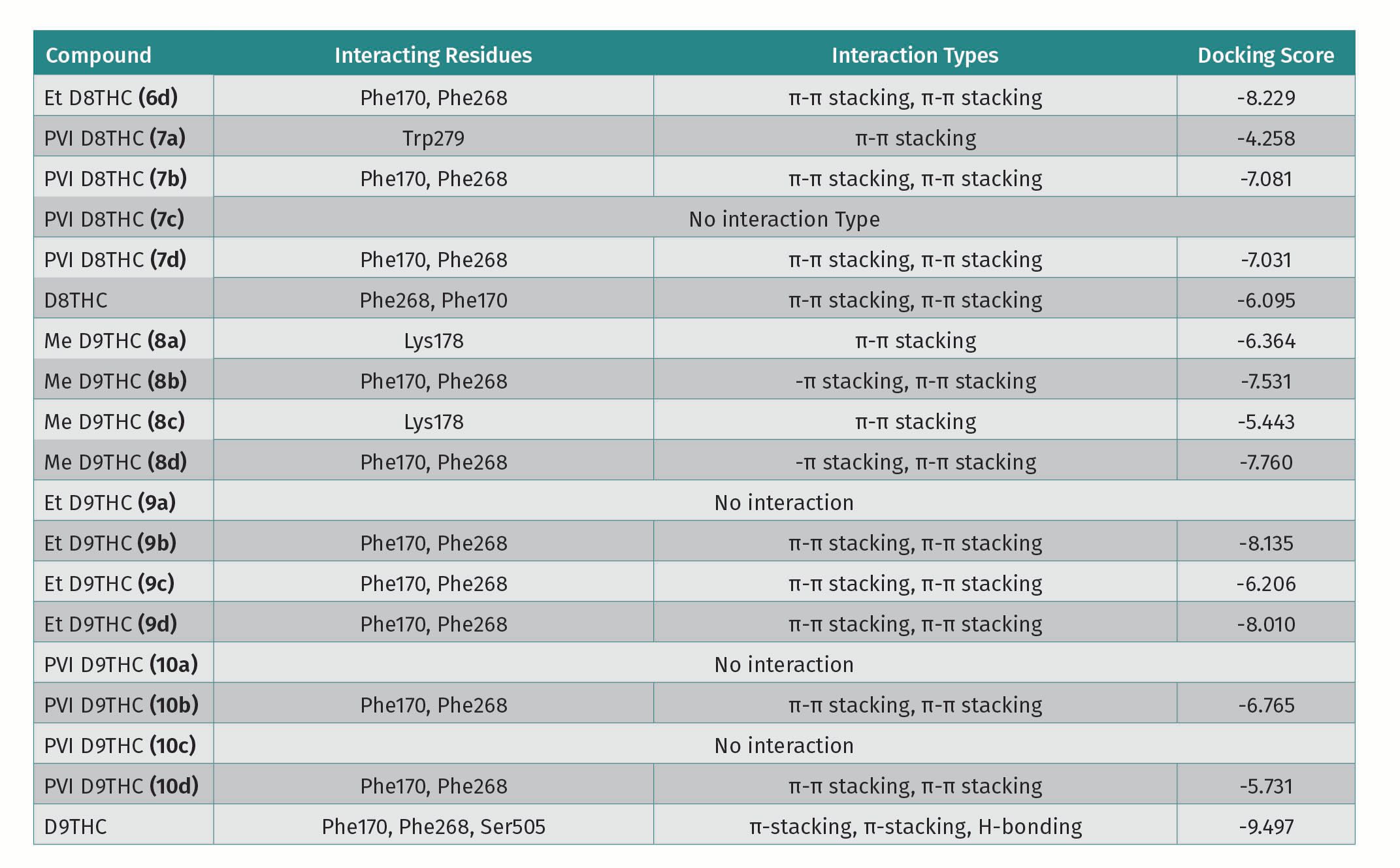
For protein 5ZTY, which is bound with AM10257 an antagonist of the CB2 receptor, the results show that all docked cannabinoid ethers interacted with the amino acid residues of this protein (Table 1), and the most common residues were Phe183 and Phe87 with π-π stacking interaction. The best docking score is -10.770 kcal/mol which corresponds to S-Me-HHC (3a) and the worst is -8.685 kcal/mol for Et-D8-THC (6d) (Table 1). In addition, Me-HHC (3a and 3h) were the only compounds that exhibited an interaction with Trp194, which is a key residue in the binding pocket of the CB2 receptor. It is interesting to point out that the R-HHC (3d, 4d) ethers and R-HHC did not display any interaction with the amino acids of the 5ZTY protein. Also, D8-THC methyl ether (5a) showed a good docking score (-10.167 kcal/mol), meanwhile, D9-THC methyl ether (8a) did not exhibit any interaction. For protein 7V3Z as a CB1 receptor with a negative allosteric modulator ORG27569 bound, the specific interactions among the docked cannabinoids and 7V3Z residues and the docking scores are disclosed in Table 1. The most important amino acids found in the binding pocket that interact with cannabinoids include Phe170 and Phe268 via π-π stacking. Most of the docked compounds displayed good docking scores between -8.561 (R-Me-HHC-compound 3d) and -4.174 (Et-D8-THC-compound 6a). It is remarkable to observe that the best docking scores were obtained for protein 5ZTY (CB2 receptor) compared to protein 7V3Z (CB1 receptor). We selected S-HHC methyl ether (3a) and D8-THC methyl ether (5a) as the compounds that revealed the best docking affinity for the CB2 receptor.
The use of in-silico software to identify the binding affinities allows us to hypothesize the effects of compounds prior to running in-vitro or in-vivo experiments to determine their physiological or behavioral effects. Experiments done from 1991-1992 using radioligand assays to determine effects of methyl ether cannabinoids, D9-tetrahydrocannabinol methyl ether (D9-THCME) was shown to have a Ki value of >10,000nM meaning the compound did not bind to CB1 receptor unless a large concentration was taken. The reference ligand to test the affinity was 3H-CP-55,940, a ligand with high affinity for CB1 and CB2 at 0.58nM and 0.68nM respectively. The ligand is used in in-vivo experiments to mimic the effects of THC (14). Behavioral experiments were performed using D8-tetrahydrocannabinol methyl ether (D8-THCME), D9-THCME, and D8-THC. D8-THCME from the invitro studies had a 3200nM binding affinity with the tail flick and behavioral characteristics being completely diminished compared to the weaker D8-THC (15). The study also acknowledges that through the use of rat and mouse testing the D8-THCME and D9-THCME are inactive and would need much higher doses to possibly create effects (15).
4.2. In Silico ADME Properties of Cannabinoid Ethers
Optimization of the ADME properties of the drug molecule is often the most difficult and challenging part of the whole drug discovery process. The ADME profile will also have a major impact on the likelihood of success of a drug. The drug-likeness and physiochemical properties of cannabinoid ethers were analyzed via Maestro’s QikProp Schrödinger software (11). The predicted ADMET properties and descriptors for the compounds are presented in Table 2. The aqueous solubility (QPlogS) is critical for the estimation of absorption and distribution of the compounds within the body and ranges between -7.150 and -9.702. These values were out of the recommended range and had poorer solubility than their analogs with the free OH group. The solubility of cannabinoids is a challenge due to their lipophilic character. QPlog-hERG is another parameter that is out of the recommended range for D8-THC and D9-THC ethers. It is predicted IC50 value for blockage of hERG K channels. Most other descriptors are within the recommended range by QikProp for 95% of known oral drugs. These results suggest that HHC-ethers exhibited acceptable physiochemical properties, however, D8-THC and D9-THC ethers can induce cardiotoxicity and result in arrhythmia (16-18), according to the hERG (Human Ether-a-go-go related gene) scores. Studies have been published showing HHC can cause a failed hERG but pass Nav/Cav, meaning although inhibition is reported of the hERG pathway there is no inhibition of the ion channels allowing for regular beating of the heart. CBD also has shown to cause issues with hERG and can cause arrhythmia and cardiac adverse events, with the use of Schrödinger we can identify possible issues based off the structure of the compound and known medicinal chemistry principles with a score of -5 or higher possibly be concern for failing hERG and the need for further testing.


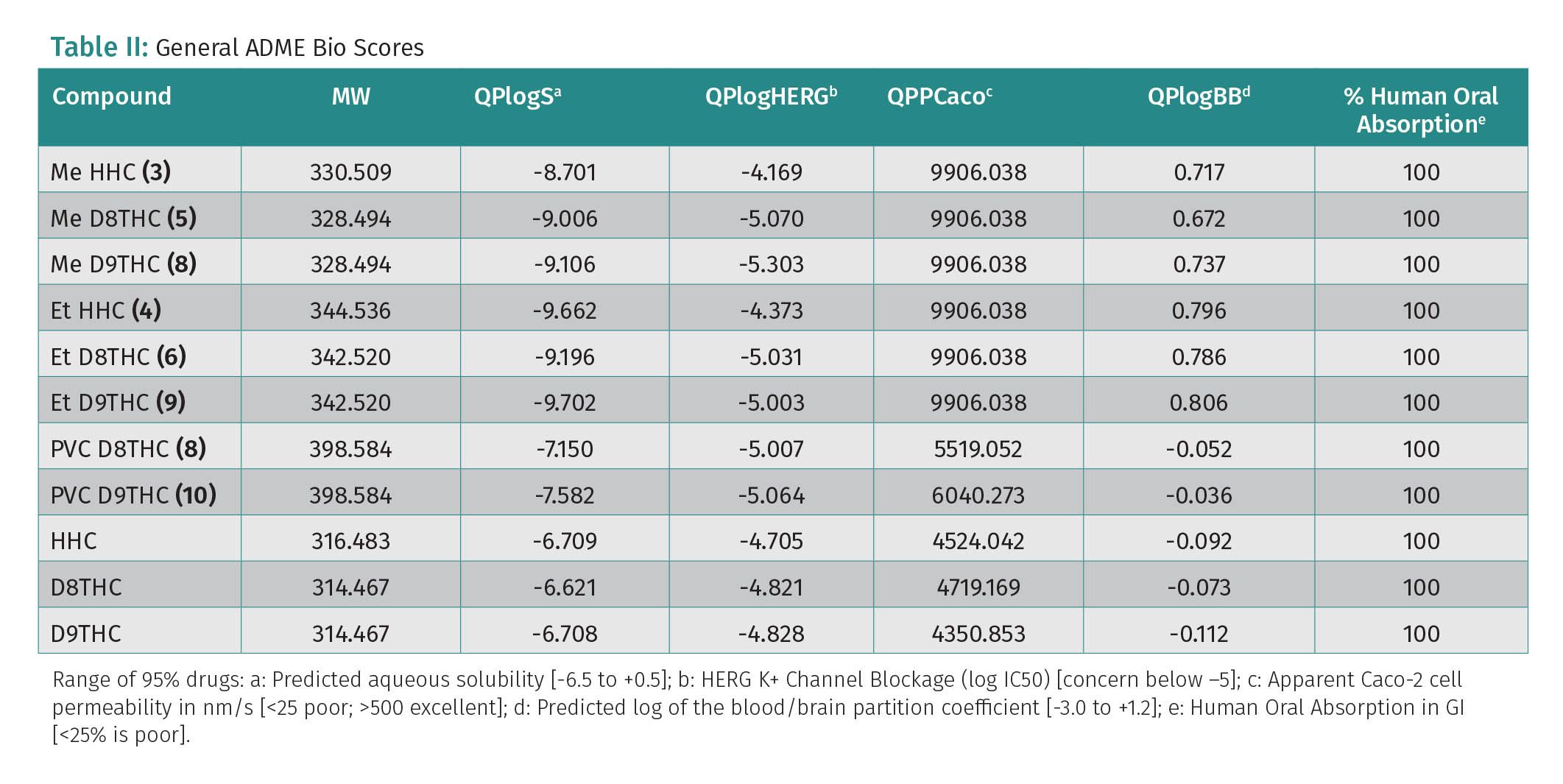
The predicted solubilities of the cannabinoids are well outside the calculated ranges, which proves that the cannabinoids are extremely lipophilic with no feasible solubility in water. The decreased solubility reduces the absorption, digestion, and metabolism of the compound if taken as pure compound with the bioavailability scores being abysmal (<5%). In studies the uptake of cannabinoids in fats, increases the bioavialability and affecting the absorption, digestion and metabolism of the cannabinoid, in an increased manner (19).
4.3 Synthesis and Characterization of HHC-Me and D8THC-Me
Our group was able to synthesize the methyl ether of HHC, starting with 6,9-dimethyl-3-pentyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromen-1-ol (11) using methyl iodide as methylation agent, and sodium hydride as base in tetrahydrofuran (THF) to obtain 1-methoxy-6,6,9-trimethyl-3-pentyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromene (3) with excellent yields (Figure 1b-c).
HHC (11), the starting material, and HHC-OMe (4), the finished product, can be identified via 1H-NMR and 13C-NMR. 1H-NMR: In the region between 3.5-4.0 ppm, compound 3 shows a singlet corresponding to the hydrogens of the methoxy group. This peak does not appear at the 1H-NMR spectrum of HHC. 13C NMR: For Me-HHC (3), in the region between 50-60 ppm appears a peak corresponding with the methyl carbon in the methoxy group, which does not appear in the HHC (11). Table 2 provides NMR spectroscopic data for the Me-HHC (4) and D9-THC-ME (5).



Our group also synthesized the methyl ether of THC, starting with 6,6,9-trimethyl-3-pentyl-6a,7,8,10a-tetrahydro-6H-benzo[c]chromen-1-ol (12) using the procedure mentioned above to obtain 1-methoxy-6,6,9-trimethyl-3-pentyl-6a,7,8,10a-tetrahydro-6H-benzo[c]chromene (5) with excellent yields. THC (12), the starting material, and Me-D8THC (5), the finished product, could be identified by 1H-NMR and 13C-NMR. 1H-NMR: At 3.8 ppm, compound 5 shows a singlet corresponding to the hydrogens of the methoxy group. This peak does not appear at the 1H-NMR spectrum of THC. 13C-NMR: For Me-D8THC (5), in the region between 55-56 ppm appears a peak corresponding with the methyl carbon in the methoxy group, which does not appear in the THC (12). Table 2 above provides NMR spectroscopic data for the Me-D8THC (5).
5. Conclusion
Our team synthesized both the THC and HHC methyl ethers in high yield using NMR to verify the addition of the methyl ether, since HPLC can cause false identification of desired compound. Molecular docking studies show that the cannabinoid methyl ethers bind favorably to CB2 over CB1 similar to the literature. The best docking affinities were shown by S-HHC methyl ether and D8-THC methyl ether. ADME results show that these compounds have low aqueous solubility and a potential for hERG K+ Channel Blockage. The production of this data provides an in-silico assessment of the binding interactions of these pro-drug moiety analogs and an importance in identifying potential issues that would need further testing to ensure safety for consumers such as the hERG values which are pertinent in the pharmaceutical fields in whether or not drugs can be safe to use.
Acknowledgments
W.C. gratefully acknowledges NMR spectroscopy support from Jin Hong at Custom NMR Services, Inc. from Woburn, MA.
References
- Durán-Iturbide, N. A.; Díaz-Eufracio, B. I.; Medina-Franco, J. L. In Silico ADME/Tox Profiling of Natural Products: A Focus on BIOFACQUIM. ACS Omega 2020, 5 (26), 16076–16084. https://doi.org/10.1021/acsomega.0c01581.
- Paul Gleeson, M.; Hersey, A.; Hannongbua, S. In-Silico ADME Models: A General Assessment of Their Utility in Drug Discovery Applications. Curr. Top. Med. Chem. 2011, 11 (4), 358–381. https://doi.org/10.2174/156802611794480927.
- Criscuolo, E.; De Sciscio, M. L.; Fezza, F.; Maccarrone, M. In Silico and in Vitro Analysis of Major Cannabis-Derived Compounds as Fatty Acid Amide Hydrolase Inhibitors. Molecules 2020, 26 (1), 48. https://doi.org/10.3390/molecules26010048.
- Docampo-Palacios, M. L.; Ramirez, G. A.; Tesfatsion, T. T.; Okhovat, A.; Pittiglio, M.; Ray, K. P.; Cruces, W. Saturated Cannabinoids: Update on Synthesis Strategies and Biological Studies of These Emerging Cannabinoid Analogs. Molecules 2023, 28 (17), 6434. https://doi.org/10.3390/molecules28176434.
- Matter, H.; Schmider, W. In-Silico ADME Modeling. In Drug Discovery and Evaluation: Safety and Pharmacokinetic Assays; Springer Berlin Heidelberg: Berlin, Heidelberg, 2013; pp 1005–1052.
- Bow, E. W.; Rimoldi, J. M. The Structure–Function Relationships of Classical Cannabinoids: CB1/CB2 Modulation. Perspect. Medicin. Chem. 2016, 8, PMC.S32171. https://doi.org/10.4137/pmc.s32171.
- Kruger, D. J.; Bone, C. C. B.; Meacham, M. C.; Klein, C.; Kruger, J. S. THC-O-Acetate: Scarce Evidence for a Psychedelic Cannabinoid. J. Psychoactive Drugs 2023, 1–5. https://doi.org/10.1080/02791072.2023.2230573.
- Khanolkar AD, Palmer SL, Makriyannis A. Molecular probes for the cannabinoid receptors. Chem Phys Lipids. 2000; 108 (1–2):37–52. http://dx.doi.org/10.1016/s0009-3084(00)00186-9
- Han, S.; Thatte, J.; Buzard, D. J.; Jones, R. M. Therapeutic Utility of Cannabinoid Receptor Type 2 (CB2) Selective Agonists. J. Med. Chem. 2013, 56 (21), 8224–8256. https://doi.org/10.1021/jm4005626.
- Docampo-Palacios, M. L., Ramirez, G. A., Tesfatsion, T. T., Pittiglio, M. K., Ray, K. P., Cruces, W., In Silico ADME, binding affinities, and properties of hydrogenated cannabinoids, Invited Submission to Pharmaceuticals (MDPI) 2023. https://doi.org/10.26434/chemrxiv-2023-7jxvh
- Schrödinger, LLC . Schrödinger Release 2021-4: QikProp. Schrödinger, LLC; 2021.
- Grant, B. J.; Gorfe, A. A.; McCammon, J. A. Large Conformational Changes in Proteins: Signaling and Other Functions. Curr. Opin. Struct. Biol. 2010, 20 (2), 142–147. https://doi.org/10.1016/j.sbi.2009.12.004
- Guan, L.; Yang, H.; Cai, Y.; Sun, L.; Di, P.; Li, W.; Liu, G.; Tang, Y. ADMET-Score – a Comprehensive Scoring Function for Evaluation of Chemical Drug-Likeness. Medchemcomm 2019, 10 (1), 148–157. https://doi.org/10.1039/c8md00472b
- Compton, D. R.; Rice, K. C.; De Costa, B. R.; Razdan, R. K.; Melvin, L. S.; Johnson, M. R.; Martin, B. R. Cannabinoid Structure-Activity Relationships: Correlation of Receptor Binding and in Vivo Activities. J. Pharmacol. Exp. Ther. 1993, 265 (1), 218–226.
- Compton, D. R.; Prescott, W. R., Jr; Martin, B. R.; Siegel, C.; Gordon, P. M.; Razdan, R. K. Synthesis and Pharmacological Evaluation of Ether and Related Analogs of .DELTA.8-, .DELTA.9-, and .DELTA.9,11-Tetrahydrocannabinol. J. Med. Chem. 1991, 34 (11), 3310–3316. https://doi.org/10.1021/jm00115a023
- Orvos, P.; Pászti, B.; Topal, L.; Gazdag, P.; Prorok, J.; Polyák, A.; Kiss, T.; Tóth-Molnár, E.; Csupor-Löffler, B.; Bajtel, Á.; Varró, A.; Hohmann, J.; Virág, L.; Csupor, D. The Electrophysiological Effect of Cannabidiol on hERG Current and in Guinea-Pig and Rabbit Cardiac Preparations. Sci. Rep. 2020, 10 (1). https://doi.org/10.1038/s41598-020-73165-2
- Garrido, A.; Lepailleur, A.; Mignani, S. M.; Dallemagne, P.; Rochais, C. hERG Toxicity Assessment: Useful Guidelines for Drug Design. Eur. J. Med. Chem. 2020, 195 (112290), 112290. https://doi.org/10.1016/j.ejmech.2020.112290
- A. Collins, T. Tesfastion, G. Ramirez, K. Ray, and W. Cruces, Cannabis Science and Technology®, 2022. 5 (7), 23-27.
- Perucca, E.; Bialer, M. Critical Aspects Affecting Cannabidiol Oral Bioavailability and Metabolic Elimination, and Related Clinical Implications. CNS Drugs 2020, 34 (8), 795–800. https://doi.org/10.1007/s40263-020-00741-5
About the Authors
Westley Cruces co-founded Colorado Chromatography Labs and joined as their Chief Science Officer in the spring of 2020. Together with Kyle Ray, he commercialized HHC manufacturing, built out multiple laboratories, returned 100% ROI within 18 months, and has authored over 10 publications and patents while at CCL. After screening various compounds against cancer cell lines, he had two hits with solid IC-50 values. The CCL team spun off BlackStone Therapeutics and set out to complete the preclinical studies needed to apply to Phase 1 Clinical Trials. Direct correspondence to wes@coloradochromatography.com.
Maite L. Docampo-Palacios has developed technologies applying the regioselective catalysis as a cost-effective alternative for the commercial manufacture of cannabinoids such as CBD and THC, as well as the rare cannabinoids CBDV, THCV, CBDP, THCP, CBN, CBC, CBT, and many more under mild conditions. She successfully brought more than 10 synthetic cannabinoids to market for the nutraceutical industry. Now, she is the Lab Director of Colorado Chromatography Labs. Her role includes leading the R&D team through the entire cycle of cannabinoids and research, development, and production from designing and optimizing the total and partial synthesis of rare cannabinoids, feasibility assessment, risk analysis, and process validation to large-scale production with low cost.
Tesfay T. Tesfatsion has worked on many CDMO projects delivering final products in a timely manner. Including validating final compounds with COA and full characterizations as needed. Currently, Tesfay is a senior scientist at Sunflower Wellness, formerly Colorado Chromatography, developing novel methods and synthesizing cannabinoids from bench to pilot scale. Including closely working the chief scientific officer in in reaching production goals and processes.
Giovanni A. Ramirez is currently an R&D chemist at Sunflower Wellness, formerly Colorado Chromatography, where he focuses mainly on computational chemistry and medicinal chemistry while also assisting in organic bench work. He also participates in writing manuscripts, patents, and technical reports.
How to Cite this Article
Docampo-Palacios, M. L., Ramirez, G. A., Tesfatsion, T. T., Cruces, W. Cannabinoid Ethers:Identification, Binding Interactions, and their ADME Properties, Cannabis Science and Technology, 2024, 7(3), 30-39.






















